Inhaltsverzeichnis
Lancaster Syndrom gibt es das wirklich?
Eine Erkrankung die sich anhört wie ein Science-Fiction-Roman? Oder ein Ritter aus dem Mittelalter? Zugegeben der Name klingt schon sehr erfunden, zumal kaum etwas dazu im Internet oder in Fachbüchern und -zeitschriften zu lesen ist. Und wenn doch, so sind die Informationen sehr spärlich gesät. Wir haben für Sie die wichtigsten Informationen zusammengetragen und wollen das Geheimnis um diese seltsame Erkrankung hier lüften.
Lancaster Syndrom was ist das – Definition?
Das Lancaster Syndrom, medizinisch besser bekannt unter dem Namen
Ellis-van-Creveld-Syndrom, ist eine sehr seltene Krankheit. Sie wird autosomal-rezessiv vererbt und gehört zu den Kurzripp-Polydaktylie-Syndromen. Der Begriff Kurzripp-Polydaktylie-Syndrom umfasst mehrere verschiedene Krankheiten, deren Hauptmerkmale kurze Rippen, ein eingeengter Brustkorb und somit unterentwickelte Lungen sind. Sie sind tödlich, wenn keine Behandlung durchgeführt wird.
Weltweit werden schätzungsweise 1 : 60000 bis 1 : 200000 Neugeborene mit dieser Erkrankung geboren. Es gibt jedoch einige Sippen wie die Aborigines oder die Amischen, bei denen eine signifikante Häufung auftritt. So liegt die Häufigkeit bei den Amischen in Lancaster County pro Neugeburt bei ca. 1 : 5000. Der Name Lancaster Syndrom wird hiervon abgeleitet.
Erstmals wurde die Erkrankung durch Richard Ellis (1902–1966) und Simon van Creveld (1894–1971), beides Pädiater, definiert. Sie definierten die Symptome dreier Patienten im Jahre 1940. Es gibt aber auch frühere Veröffentlichungen, welche die Erkrankung beschreiben, jedoch nicht definieren.
Im Folgenden finden Sie die wichtigsten Informationen zum Ellis-van-Creveld-Syndrom.
Welche Auswirkung hat das Lancaster Syndrom?
Genetik und Pathologie
Wie bereits erwähnt, ist diese Erkrankung vererbt. Die Ursache ist vermutlich in einer Mutation der Gene EVC1 und EVC2 zu finden. Diese Gene liegen nebeneinander auf dem kurzen Arm des Chromosoms 4 und haben ähnliche Aufgaben. Die Forschung ist hier aber bei Weitem noch nicht abgeschlossen und eine eindeutige Lancaster Syndrom Erklärung lässt sich bis heute nicht finden.
Die Weitervererbung kann nur eines der Gene betreffen, d. h. die Kinder erhalten von beiden Elternteilen jeweils das gleiche mutierte Gen, beispielsweise EVC1. Die Kinder können aber auch vom einen Elterteil EVC1 erben und vom anderen EVC2. Zusätzlich wird angenommen, dass weitere Mutationen oder Gendefekte an der Erkrankung beteiligt sind. Unterschiede in der Erkrankung sind allerdings keine zu finden. Die Symptome äußern sich bei allen Betroffenen gleich.
Defekte auf Chromosom 4 können aber auch zu anderen genetisch vererbten Erkrankungen führen wie etwa das Weyers-Syndrom.
Die Genetik und Pathologie der Erkrankung sind sehr komplex. Untersuchungen zeigten, dass etwa 70 % der, zum Verständnis der Krankheit abgeklärten, Patienten eine Mutation auf den genannten Genen EVC1 oder EVC2 aufweisen. Die restlichen 30 % weisen jedoch keine Mutationen auf diesen Genen auf.
Welche Ursachen gibt es für das Syndrom?
Ist das Lancaster Syndrom eine Nervenkrankheit?
Wie es zu einer Mutation an den genannten Genen EVC1 und EVC2 kommt, ist weitgehend unbekannt. Eine allgemein gültige Lancaster Syndrom Erklärung die begründen könnte, welche Auslöser dafür verantwortlich sind, gibt es nicht. Allerdings fällt auf, dass bei der Mehrzahl aller Erkrankten ein falsch gesetztes Stopcodon die Krankheit ausgelöst hat. Ein Stopcodon ist ein Bestandteil der DNA, bzw. der RNA.
Aber durch die Tatsache, dass in einigen Fällen keine Veränderungen auf diesen beiden Gene nachgewiesen werden konnten, macht die Frage nach der Ursache sehr schwierig.
Lancaster Symptome
Die Erkrankung zeichnet sich durch verschiedene charakteristische Merkmale aus. Die Betroffenen leiden unter ektodermalen Defekten und Herzfehlern, kurzen Rippen, an Minderwuchs und Polydaktylie (Vielfingerigkeit).
Äußerlich fällt dementsprechend der Minderwuchs und eine Verformung des Brustkorbes mit verkürzten Rippen auf. Der Minderwuchs ist ein typisches Zeichen. Er entsteht zwangsläufig durch verkürzten Gliedmaßen. Die effektive Größe im Erwachsenenalter ist sehr schwer vorauszusagen.
Meistens besitzen die Betroffenen einen sechsten Finger an jeder Hand. Dieser befindet sich neben dem kleinen Finger und ist nach außen gerichtet. Die Polydaktylie betrifft in 90 % der Fälle nur die Hände. Nur gerade etwa 10 % der Betroffenen besitzen auch an den Füssen eine überzählige Zehe. In vielen Fällen lässt sich jedoch ein vergrößerter Zwischenraum zwischen der Großzehe und der zweiten Zehe finden.
Weiter leiden die Patienten unter verkürzten Gliedmassen, sowohl der Arme und Beine, als auch der Finger und Zehen. Oft ist es ihnen daher nicht möglich die Hand zu einer Faust zu schließen.
Auch eine chondrale und ektodermale Dysplasie sowie Veränderungen am Herzen im Sinne eines Atriumseptumdefektes werden häufig beschrieben. Die chondrale und ektodermale Dysplasie betreffen hier das Skelettwachstum und Fehlbildungen an Nägel und Zähnen. Beispielsweise können Zähne fehlen oder veränderte Formen ausbilden, wie etwa konisch anstatt typisch zahnförmig. Auch treten gelegentlich Gaumenspalten auf.
Anzeichen
Die äußerlichen Anzeichen wie die Polydaktylie oder die Verformung des Brustkorbes werden spätestens bei der Geburt offensichtlich. Aber auch während der Schwangerschaft können gegen Ende des ersten Trimester mittels einer Pränataldiagnostik bereits verschiedene Symptome erkannt werden. Ein Feinultraschall kann einen verengten Thorax, Verkürzungen der langen Röhrenknochen in Armen und Beinen, eine Hexadaktylie (Sechsfingrigkeit) oder Herzfehler aufzeigen. Da aber auch andere Erkrankungen ähnliche Symptome hervorrufen, müssen diese differentialdiagnostisch von einem Lancaster Syndrom abgegrenzt werden.
Diagnose
Die Diagnose wird durch die Pränataldiagnostik während der Schwangerschaft oder bei einer körperlichen Untersuchung durch Inspektion anhand der offensichtlichen Symptome gestellt. Ein Gentest EVC1 und EVC2 ist ebenfalls möglich um eine sichere Diagnose stellen zu können.
Weitere Untersuchungen können auch radiologischer Natur sein. Mittels Röntgen, einer Magnetresonanztomographie (MRT) oder einer Computertomographie (CT) können verschiedene Kriterien untersucht werden.
- Beckendysplasie mit Spornbildung medial am Pfannendach
- Überproportional kurze Unterarme und Unterschenkel
- Verbreiterung der Tibia proximal, nach medial verlagertes Ossifikationszentrum mit massivem Genu valgum
- Verkürzte Fingerglieder mit Zapfenepiphysen der Mittel- und Endphalangen
- Vorzeitige Ossifikation der proximalen Oberarm- und Oberschenkelepiphyse
- Zu kurze Fibula
Komplikationen
Die Komplikationen sind häufig psychischer Natur. Durch die Fehlbildungen und das äußere Erscheinungsbild leiden die Betroffenen oftmals unter Mobbing und einem verringerten Selbstwertgefühl.
Besteht ein Herzfehler so kann dies zu einer verringerten Lebenserwartung führen.
Meist leiden die Betroffenen durch den deformierten Brustkorb auch unter Atmungsproblemen.
Keinen Einfluss hat die Erkrankung auf die geistige Entwicklung der Betroffenen. Das Denken und die Intelligenz sind nicht eingeschränkt.
Lancaster Syndrom Lebenserwartung
Die Lebenserwartung des Lancaster Syndroms ist abhängig von den Missbildungen des Brustkorbes und des Atriumseptumdefektes oder anderen Herzfehlbildungen. Sie hängt auch zusammen mit den eingeleiteten Therapiemaßnahmen.
Grundsätzlich kann die Lebenserwartung gut erhöht werden. Dazu ist aber eine konsequente Behandlung notwendig.
Therapie
Das Lancaster Syndrom ist bis heute noch nicht heilbar.
Die Therapie richtet sich demnach nach den Symptomen. Es wichtig, dass die Patienten schon ab der Geburt eine fachärztliche Betreuung erhalten. Regelmäßige Kontrollen bei den verschiedenen Fachärzten sind notwendig um rechtzeitig Maßnahmen ergreifen zu können. In erster Linie soll die Überwachung und Therapie der vital gefährdenden Symptome wie Herzfehler und Dyspnoe behandelt werden. Je nach dem können auch orthopädische Eingriffe die Knochendeformitäten korrigieren.
Weiter sind symptomspezifische alternative Möglichkeiten vorhanden. Verschiedene Therapieansätze sollen dabei die Beschwerden der Betroffenen so angenehm wie möglich machen.
- Physio- und Ergotherapie
In der Physiotherapie oder Ergotherapie lernen die Patienten ihre Bewegungsmöglichkeiten, bspw. Handbewegungen, optimal auszunutzen. Die Körperhaltung ist sehr wichtig für die Atmung. Ebenso können mit der richtigen Körperhaltung Schmerzsyndromen entgegengewirkt werden.
- Atemtherapie
Die Deformierung des Thorax verlangt nach besonderen Atemtechniken. Daher ist eine Atemtherapie angebracht um die Beherrschung der Atmungs und ihrer besonderen Schwierigkeiten zu erlernen.
- Zähne
Die Besonderheiten der Zähnen können verschiedene Komplikationen nach sich ziehen. Eine regelmäßige Kontrolle bei einem Zahnarzt sollte daher jährlich durchgeführt werden, bei Bedarf auch mehrmals jährlich.
- Psychotherapie
Da die Patienten oft an einem Minderwertigkeitskomplex, verringertem Selbstvertrauen und verschiedenen Ängsten leiden, ist eine Psychotherapie oder eine psychologische Begleitung manchmal unumgänglich. Mobbing, bspw. durch nicht erkrankte Geschwister, im weiteren Umfeld und Ablehnung durch die Gesellschaft hinterlässt bei vielen negative Gefühle. Die Betroffenen können ein selbstzerstörerisches Bild ihrer Person entwickeln oder ziehen sich aus der Gesellschaft zurück. Aber auch scheinbar psychisch gesunde Patienten sind nicht immer stabil. Durch eine gezielte psychotherapeutische oder psychologische Begleitung werden die Betroffenen aufgebaut. Sie erlernen Strategien und können ein gesundes Selbstvertrauen aufbauen.
- Medikamente
Medikamente, welche einen direkten Einfluss auf den Gendefekt haben, gibt es keine. Je nach Beschwerdebild, ist es aber möglich verschiedene Medikamente einzunehmen.
Bei Atemproblemen oder einer möglichen Asthmaproblematik können Inhaler verschrieben werden.
Psychopharmaka werden bei Bedarf bei psychologischen Problemen eingesetzt. Dies können Stimmungsaufheller oder spannungslösende Produkte sein, aber auch Schlaftabletten bei Schlafstörungen.
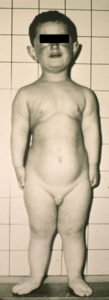
see above, Ellis-vancreveld, CC BY 2.0
Vorbeugung
Der Erkrankung kann nicht vorgebeugt werden. Es ist allerdings sinnvoll eine genetische Beratung durchführen zu lassen, wenn bereits ein Diagnose in der Familie vorliegt. Da die Erkrankung aber autosomal rezessiv weitergegeben wird, ist das Risiko einer Erkrankung sehr gering. Eine Prognose einer möglichen Erkrankung ist sehr schwer abzugeben.
Differentialdiagnosen
Hier werden verschiedene ähnliche Erkrankungen kurz vorgestellt.
Asphyxierende Thoraxdysplasie
Die asphyxierende Thoraxdysplasie, auch unter dem Namen Jeune-Syndrom bekannt, ist eine häufig letale Skelettdysplasie. Sie gehört ebenfalls zu den Kurzripp-Polydaktylie-Syndromen.
Auch wenn sie ebenfalls autosomal rezessiv vererbt wird ist ihre Ursache nicht in den Genen EVC zu finden. Die Häufigkeit wird hier mit 1-5 Erkrankte auf 500 000 Personen angegeben.
Die Symptome sind fast die gleichen wie beim Ellis-van-Creveld-Syndrom, daher ist eine Abgrenzung dazu sehr schwierig.
McKusick-Kaufman-Syndrom
Hierbei handelt es sich um ein Syndrom mit angeborenen Fehlbildungen der Genitalien (Atresie des Ureters, Doppelung von Vagina und/oder Uterus, Hydrometrokolpos, Hypospadie, Ureterstenose, ), postaxialer Polydaktylie und Herzfehlern. Eine Häufigkeit kann mit 1 % der Geburten bei den Amischen angegeben werden. Ansonsten sind keine Angaben über die Häufigkeit vorhanden.
Die Ursache lässt sich auf Mutationen im MKKS-Gen finden.
Pallister-Hall-Syndrom
Dieses angeborene Syndrom geht mit Fehlbildungen im Gehirn (Hamartom im Hypothalamus, Hypopituitarismus), Analatresien und postaxialen Polydaktylien einher.
Es handelt sich um eine autosomal dominant vererbte Erkrankung. Insgesamt sind weltweit etwa 100 Fälle bekannt.
Mutationen im GLI3-Gen sind für die Fehlbildungen verantwortlich.
Verma-Naumoff-Syndrom
Das Verma-Naumoff-Syndrom, auch bekannt als Kurzrippen-Polydaktylie-Syndrom Typ 3, ist eine angeborene vererbare Krankheit mit Osteochondrodysplasien mit letalem Verlauf. Kurze Rippen und eine unterentwickelte Lunge mit Ateminsuffizienz sind die Hauptmerkmale. Hinzu kommen verschiedene komplexe Fehlbildungen, bspw. Analatresien Herzfehler, Genitalhypoplasie, Kurzdarm, Malrotation, Nierenhypoplasien oder -aplasien, Urethralatresie.
Die Erkrankung ist tödlich.
Diese Erkrankungen sind klar von einem Ellis-van-Creveld Syndrom abzugrenzen um eine korrekte Diagnose stellen zu können. Die Schwierigkeit liegt darin, dass die Erkrankungen auf den ersten Blick einige gleiche Symptome aufweisen. Hier genau hinzuschauen ist daher besonders wichtig.
Wo und in welcher Form erscheint das Lancaster Syndrom?
Das Lancaster Syndrom tritt besonders häufig bei den Amischen in Lancaster County und bei den Aborigines auf. Bis zum Jahr 2000 wurden bei den Amischen in 30 Sippen insgesamt 52 Fälle registriert. Weltweit gibt es verschiedene Berichte mit einer Gesamtzahl von 150 bis 300 Erkrankten.
Unklar ist es, wieso ausgerechnet bei den Amischen oder den Aborigines gehäuft Fälle zu beobachten sind. Die Frage liegt Nahe, ob diese Gendefekte etwas mit früherem Inzest zu tun haben. Aufzeichnungen darüber sind jedoch keine zu finden.
Auch in Nord- und Südamerika wurden Mutationen des EVC-Genes entdeckt. In welchen Gruppen diese Mutationen beschrieben wurden, ist aber nicht öffentlich dokumentiert.
Außerdem sind die übrigen 100 bis 250 bekannten Fälle in verschiedenen Gebieten der Welt zu finden.
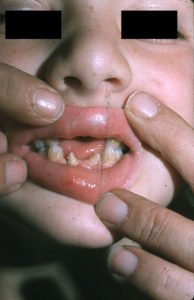
Baujat G, Le Merrer M., Mouth ECS, CC BY 2.0
Die Form der Erkrankung ist bei allen grundsätzlich gleich. Variieren können die verschiedenen äußerlichen Symptome; das heißt die Polydaktylie, die Verformungen an Zähnen und Nägeln oder die Gaumenspalte.
Die oben genannten Symptome und Anzeichen können unterschiedlich stark ausgebildet sein. Sie betreffen Arme und Beine, Finger und Zehen, das Gebiss und den Mund sowie (hauptsächlich) den Thorax und das Herz wie folgt:
- Ektodermale Defekte und Herzfehler
- Fehlende Zähne oder in der Form veränderte Zähne
- Kurze Rippen und ein deformierter Brustkorb
- Minderwuchs
- Polydaktylie mit möglichen hypo- oder dysplastischen Fingernägel
Es muss hier nochmals erwähnt werden, dass es sich um eine reale, nicht erfundene Erkrankung handelt.
Erfahrungen von Erkrankten
Die Krankheit ist sehr selten. Verschiedene Länder bieten Patientennetzwerke für Betroffene. Diese Netzwerke bieten jedoch nichts speziell auf das vorliegende Syndrom an. So gibt es in Deutschland bspw. eine Gruppierung für Patienten mit ektodermalen Dysplasien. Diese sind beim Lancaster Syndrom jedoch nur ein Symptom von mehreren.
Es lassen sich auch keine Erfahrungberichte in den Medien oder im Internet finden.
Erfahrungsberichte sind auch deshalb sehr schwer ausfindig zu machen, weil die Betroffenen häufig in spezielle Gruppen (Aborigines, Amische) hineingeboren werden oder aber vermutlich eine große Angst vor einer Stigmatisierung vorherrscht. Sie gehen mit ihrer Erkrankung nur ungern an die Öffentlichkeit.
Während Berichte sehr schwer zu finden sind, ist es bei Bildmaterial schon einfacher. In beiden Fällen ist es aber wichtig nicht unter dem Namen Lancaster-Syndrom zu suchen, sondern unter dem Namen Ellis-van-Creveld Syndrom.
Der Grund dafür ist, dass die Erkrankung im Internet häufig mit der Krankheit Franceschetti verwechselt wird. Dies, weil ein Fitnesstrainer mit dem Nachnamen Lancaster daran erkrankte, der sich stark für Erwachsene und vor allem Kinder einsetzt, welche ebenfalls daran erkrankt sind.
Die Franceschetti- Erkrankung ist ebenfalls eine Erbkrankheit, hat aber nichts mit dem Lancaster-Syndrom gemeinsam. Ihre Problematik ist, dass der Betroffene an Missbildungen der Gesichtsknochen leidet und der Patient so verunstalten.
FAQ – Fragen und Antworten
Lancaster Syndrom gibt es das wirklich?
Ja, definitiv. Dieses Syndrom ist zwar sehr selten, aber dass es existiert ist unbestritten.
Einige andere Erkrankungen, meist besser erforscht und um einiges bekannter, sind der Erkrankung jedoch sehr ähnlich. Deshalb ist eine Verwechslung, gerade für Laien, schnell möglich.
Zudem ist die Erkrankung so selten, dass kaum einer sie kennt. Viele sind daher der Meinung, dass es sich um eine erfundene Krankheit handelt. Dem ist jedoch nicht so. Ellis-van-Creveld-Syndrom bei Wikipedia.
Lancaster Syndrom was ist das?
Bei dieser Erkrankung handelt es sich um eine ererbte Krankheit. Aufgrund eines oder mehreren Gendefekten kommt es zu verschiedenen körperlichen Deformationen. Wie es zu diesen Genveränderungen kommt und wieso nicht alle Patienten an den genannten Genen Mutationen aufweisen, wie bisher angenommen, ist noch nicht vollständig geklärt. Patienten leiden unter einer Einengung des Brustkorbes, Defekten am Herzen und verschiedenen äußerlich sichtbaren Symptomen wie etwa der Vielfingrigkeit, Kleinwuchs oder Veränderungen am Gebiss.
Wie hoch ist die Lebenserwartung?
Die Lancaster Syndrom Lebenserwartung ist abhängig von der Ausprägung der Deformationen und einer allfälligen Herzerkrankung. Mit einer gezielten Behandlung kann die Lebenserwartung jedoch gesteigert werden. Allerdings ist die Erkrankung nicht heilbar.
Wie hoch ist die Wahrscheinlichkeit einer Erkrankung?
Weltweit leiden etwa 150 bis 300 Menschen an diesem Syndrom. Die Wahrscheinlichkeit, dass ein Ungeborenes daran erkrankt, in dessen Verwandtschaft sich bereits ein Erkrankter befindet, ist sehr gering. Eine Prognose, ob ein Kind erkrankt ist sehr schwer zu stellen, da es offensichtlich nicht zwingend eine Mutation an den Genen EVC1 und EVC2 sein muss, um daran zu erkranken.
Kann ich plötzlich daran erkranken?
Nein. Hier handelt es sich um einen angeborenen Gendefekt. Die Kinder kommen bereits mit verschiedenen Deformationen zur Welt. Diese bilden sich nicht im Verlaufe der Zeit aus. Es ist also nicht möglich, dass Ihnen plötzlich ein sechster Finger wächst und deshalb das Syndrom bei Ihnen diagnostiziert wird.
Fazit
Das es sich beim Lancaster Syndrom um einen angeborenen Gendefekt handelt, ist dieser natürlich sehr selten und leider nur schwer behandelbar. Man sollte falls jemand im Bekanntenkreis darunter leidet, einen Experten aufsuchen.